今回は前回に引き続き化合物ライブラリーのクラスタリングの方法を解説していきます。
また前回はPCAによるクラスタリングの可視化(グラフ化)を行ったのですが、今回は別方としてT-SNEによる可視化を行っていきます。
(また前回に引き続きセレックの実際に販売されている化合物ライブラリーを用いました。)
ダウンロード先 :
それでは早速コードを書いてみましょう。
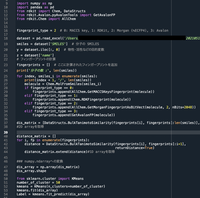
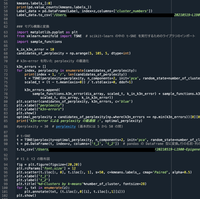
Fig. 1 Programing codes on the spyder software
長いですが、以上のようになります。前回同様、以下の方を大変参考にさせてただき少々筆者が改変したものとなります1)。
また、計算時間は1-2分ほどになりましたね。(やはり600を超えるとなかなか。。)
今回も前回同様、分子の類似性の計算方法にタニモト係数を用いました。この方法は論文などでも一般的に使われる手法であり、高い精度で比較することができます2)。
フィンガープリントはMorganの手法を用いています。
それでは早速PCAで行った場合とT-SNEで行った場合の比較をしてみましょう。
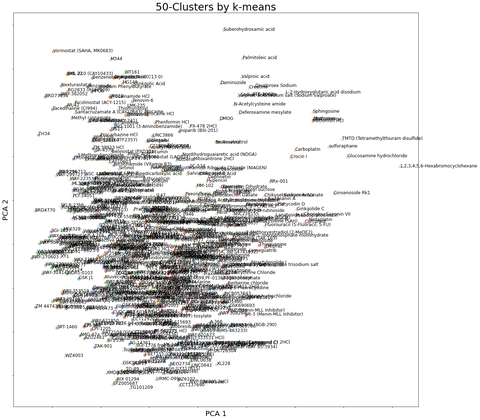
Fig. 2 Clustering of the compounds library (PCA)
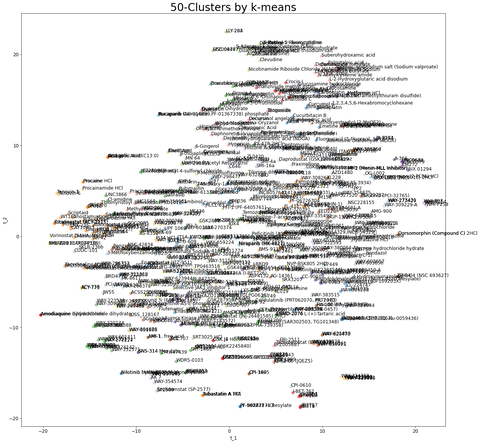
Fig. 3 Clustering of the compounds library (T-SNE)
このようになります。まずPCAは縦軸と横軸が意味を持っている軸なのに対して(タニモト係数が全体的に高い等)、T-SNEの縦軸横軸は何の意味も含まれていません。
なので、全体的に中央になるように適当にプロットされています。
今回のようにタニモト係数を用いた場合だと、タニモト係数の意味をグラフに反映させなくても良いと考えられるので、T-SNEが最適であると考えられます。
しかし、変数が少ない場合や、説明変数をうまくグラフに反映させたい場合はPCAが最適であると考えられます。
クラスタリングの方法についてはk-meansによってクラスタリングしています。この方法は最初に初期値を設定し、その初期値から近いものをグループ化していく手法になります4)。
そこでこの手法の欠点が、初期値に依存してしまうことです。
特に今回のようにデータが複雑で、クラスタリングが難しい場合は初期値の依存の問題が顕著に現れてきます。(クラスタリングの結果が1回目と2回目で変わったりします。)
次に以下でコードの解説をします5)。
【参考文献】
1) 化学のための Pythonによるデータ解析・機械学習入門、金子弘昌 著、オーム社発行、2019年出版
2) Haigh, J. A., Pickup, B. T., Grant, J. A. & Nicholls, A. Small molecule shape- fingerprints. J. Chem. Inf. Model. 45, 673–684 (2005)
3) https://future-chem.com/rdkit-fingerprint/
4) https://pythondatascience.plavox.info/scikit-learn/クラスタ分析-k-means
5) https://scikit-learn.org/stable/modules/generated/sklearn.cluster.KMeans.html
また前回はPCAによるクラスタリングの可視化(グラフ化)を行ったのですが、今回は別方としてT-SNEによる可視化を行っていきます。
(また前回に引き続きセレックの実際に販売されている化合物ライブラリーを用いました。)
ダウンロード先 :
それでは早速コードを書いてみましょう。
import numpy as np
import pandas as pd
from rdkit import Chem, DataStructs
from rdkit.Avalon.pyAvalonTools import GetAvalonFP
from rdkit.Chem import AllChem
fingerprint_type = 2 # 0: MACCS key, 1: RDKit, 2: Morgan (≒ECFP4), 3: Avalon
dataset = pd.read_excel('/Users/user's name/directory'name/20210519-L1900-Epigenetics-Compound-Library-96-well.xlsx', sheet_name=1) # SMILES 付きデータセットの読み込み
smiles = dataset['SMILES'] # 分子の SMILES
y = dataset.iloc[:, 0] # 物性・活性などの目的変数
z = dataset['name']
# フィンガープリントの計算
fingerprints = [] # ここに計算されたフィンガープリントを追加
print('分子の数 :', len(smiles))
for index, smiles_i in enumerate(smiles):
print(index + 1, '/', len(smiles))
molecule = Chem.MolFromSmiles(smiles_i)
if fingerprint_type == 0:
fingerprints.append(AllChem.GetMACCSKeysFingerprint(molecule))
elif fingerprint_type == 1:
fingerprints.append(Chem.RDKFingerprint(molecule))
elif fingerprint_type == 2:
fingerprints.append(AllChem.GetMorganFingerprintAsBitVect(molecule, 2, nBits=2048))
elif fingerprint_type == 3:
fingerprints.append(GetAvalonFP(molecule))
dis_matrix = [DataStructs.BulkTanimotoSimilarity(fingerprints[i], fingerprints[:len(smiles)],returnDistance=True) for i in range(len(smiles))]
#2D arrayを取得
distance_matrix = []
for i, fp in enumerate(fingerprints):
distance = DataStructs.BulkTanimotoSimilarity(fingerprints[i], fingerprints[:i+1],
returnDistance=True)
distance_matrix.extend(distance)#1D arrayを取得
### numpy.ndarrayへの変換
dis_array = np.array(dis_matrix)
dis_array.shape
from sklearn.cluster import KMeans
number_of_cluster = 50
kmeans = KMeans(n_clusters=number_of_cluster)
kmeans.fit(dis_array)
Label = kmeans.fit_predict(dis_array)
kmeans.labels_[:0]
print(pd.value_counts(kmeans.labels_))
Label_data = pd.DataFrame(Label, index=z,columns=['cluster_numbers'])
Label_data.to_csv('/Users/user's name/directory'name/20210519-L1900-Epigenetics-Compound-Library-96-well.csv')
### モデル構築と変換
import matplotlib.pyplot as plt
from sklearn.manifold import TSNE # scikit-learn の中の t-SNE を実行するためのライブラリのインポート
import sample_functions
k_in_k3n_error = 10
candidates_of_perplexity = np.arange(5, 105, 5, dtype=int)
# k3n-error を用いた perplexity の最適化
k3n_errors = []
for index, perplexity in enumerate(candidates_of_perplexity):
print(index + 1, '/', len(candidates_of_perplexity))
t = TSNE(perplexity=perplexity, n_components=2, init='pca', random_state=number_of_cluster).fit_transform(dis_array)
scaled_t = (t - t.mean(axis=0)) / t.std(axis=0, ddof=1)
k3n_errors.append(
sample_functions.k3n_error(dis_array, scaled_t, k_in_k3n_error) + sample_functions.k3n_error(
scaled_t, dis_array, k_in_k3n_error))
plt.scatter(candidates_of_perplexity, k3n_errors, c='blue')
plt.xlabel("perplexity")
plt.ylabel("k3n-errors")
plt.show()
optimal_perplexity = candidates_of_perplexity[np.where(k3n_errors == np.min(k3n_errors))[0][0]]
print('k3n-error による perplexity の最適値 :', optimal_perplexity)
#perplexity = 30 # perplexity (基本的には 5 から 50 の間)
# t-SNE
t = TSNE(perplexity=optimal_perplexity, n_components=2, init='pca', random_state=number_of_cluster).fit_transform(dis_array)
t = pd.DataFrame(t, index=z, columns=['t_1', 't_2']) # pandas の DataFrame 型に変換。行の名前・列の名前も設定
t.to_csv('/Users/user's name/directory'name/20210519-L1900-Epigenetics-Compound-Library-96-well_clustering_tsne_t.csv') # csv ファイルに保存。同じ名前のファイルがあるときは上書きされるため注意
# t1 と t2 の散布図
fig = plt.figure(figsize=(20,20))
plt.rcParams['font.size'] = 12
plt.scatter(t.iloc[:, 0], t.iloc[:, 1], s=50, c=kmeans.labels_, cmap='Paired', alpha=0.5)
plt.xlabel('t_1')
plt.ylabel('t_2')
plt.title('%d-Clusters by k-means'%number_of_cluster, fontsize=28)
for i, txt in enumerate(z):
plt.annotate(txt, (t.iloc[:,0][i], t.iloc[:,1][i]))
plt.show()


Fig. 1 Programing codes on the spyder software
長いですが、以上のようになります。前回同様、以下の方を大変参考にさせてただき少々筆者が改変したものとなります1)。
また、計算時間は1-2分ほどになりましたね。(やはり600を超えるとなかなか。。)
今回も前回同様、分子の類似性の計算方法にタニモト係数を用いました。この方法は論文などでも一般的に使われる手法であり、高い精度で比較することができます2)。
フィンガープリントはMorganの手法を用いています。
それでは早速PCAで行った場合とT-SNEで行った場合の比較をしてみましょう。

Fig. 2 Clustering of the compounds library (PCA)

Fig. 3 Clustering of the compounds library (T-SNE)
このようになります。まずPCAは縦軸と横軸が意味を持っている軸なのに対して(タニモト係数が全体的に高い等)、T-SNEの縦軸横軸は何の意味も含まれていません。
なので、全体的に中央になるように適当にプロットされています。
今回のようにタニモト係数を用いた場合だと、タニモト係数の意味をグラフに反映させなくても良いと考えられるので、T-SNEが最適であると考えられます。
しかし、変数が少ない場合や、説明変数をうまくグラフに反映させたい場合はPCAが最適であると考えられます。
クラスタリングの方法についてはk-meansによってクラスタリングしています。この方法は最初に初期値を設定し、その初期値から近いものをグループ化していく手法になります4)。
そこでこの手法の欠点が、初期値に依存してしまうことです。
特に今回のようにデータが複雑で、クラスタリングが難しい場合は初期値の依存の問題が顕著に現れてきます。(クラスタリングの結果が1回目と2回目で変わったりします。)
次に以下でコードの解説をします5)。
from sklearn.cluster import KMeans #ここでKMeansのライブラリをインポートしています
number_of_cluster = 50 #クラスターの数を指定しています
kmeans = KMeans(n_clusters=number_of_cluster) #指定されたクラスターの数をkmeansに設定しています
kmeans.fit(dis_array) #タニモト係数による類似性スコアをkmeansによって計算させています
Label = kmeans.fit_predict(dis_array) #クラスタリングの数やその重心を計算させています
kmeans.labels_[:0] #グループ分けしたもののナンバリングを0から今回は50までしています
print(pd.value_counts(kmeans.labels_)) #ナンバリングしたもののグループにいくつの化合物が入っているのか表示しています
Label_data = pd.DataFrame(Label, index=z,columns=['cluster_numbers']) #化合物の名前をインデックスに指定し、クラスタリングした化合物のグループ番号をカラムに指定してpandasのデータフレームを作成しています
Label_data.to_csv('/Users/ryosukenagasawa/documents/Experimental_data/FID_assay/20210519-L1900-Epigenetics-Compound-Library-96-well.csv') #作成したデータフレームをcsvファイルに変換しています

FIg. 4 Clustering excel file by means of kmeans method
結論としては、PCAとT-SNEは使い分けることが重要だと思います。
それでは今回はここまで。FIg. 4 Clustering excel file by means of kmeans method
結論としては、PCAとT-SNEは使い分けることが重要だと思います。
【参考文献】
1) 化学のための Pythonによるデータ解析・機械学習入門、金子弘昌 著、オーム社発行、2019年出版
2) Haigh, J. A., Pickup, B. T., Grant, J. A. & Nicholls, A. Small molecule shape- fingerprints. J. Chem. Inf. Model. 45, 673–684 (2005)
3) https://future-chem.com/rdkit-fingerprint/
4) https://pythondatascience.plavox.info/scikit-learn/クラスタ分析-k-means
5) https://scikit-learn.org/stable/modules/generated/sklearn.cluster.KMeans.html



コメント